SÍNDROME DE
HURLER
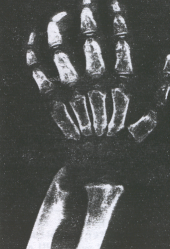
Fig. 8.17. Síndrome de Hurler. Adelgazamiento
progresivo de las falanges terminales.15
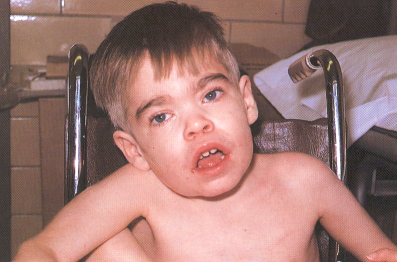
Fig. 8.18. Síndrome de Hurler. Aspecto
facial típico.1
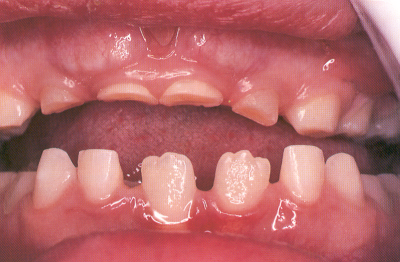
Fig. 8.19. Síndrome de Hurler.15
El síndrome de Hurler (mucopolisacaridosis
1H) es el más grave de las mucopolisacaridosis. Dado que los mucopolisacáridos
son componentes importantes de la sustancia intercelular del tejido
conjuntivo, las alteraciones óseas son características.
Las deformidades óseas que se observan en las radiografías
se denominan disostosis múltiple (Fig. 8.17). También puede
estar afectado el SNC. Se sospecha de la presencia de este trastorno
en funcion a las manifestaciones clínicas y radiológicas,
y el diagnóstico se confirma mediante la identificación
de un aumento de la excreción urinaria de mucopolisacáridos
y un déficit de la enzima específica.1
La frecuencia de mucopolisacaridosis 1H es aproximadamente 1 por 100,000 nacimientos. El defecto básico en
la enfermedad de Hunter es un déficit de alfa-L-Iduronidasa, que da
lugar a la acumulación de dermatán y heparán sulfato
en los tejidos y a su excreción por la orina.15
Están afectados casi todos los tejidos
del organismo, con una amplia aparición de células vacuoladas.
Estas células contienen lisosomas repletos de mucopolisacáridos.
En el cerebro se produce también un depósito de lípidos
junto con la acumulación de mucopolisacáridos.1
Los niños con un síndrome de Hurler
tienen un aspecto normal al nacer y durante el primer
años de vida tan solo se aprecian leves retrasos del desarrollo.
Sin embargo, a la exploración física se detecta hepatosplecnomegalia,
una cifosis excesiva, una secreción nasal persistente y una respiración
ruidosa. Las características faciales se van haciendo más
toscas a partir del primer años de
vida. La cabeza es grande y dolicocefálica, con un abombamiento frontal
y suturas sagital y metópica prominentes. El puente de la nariz
está deprimido; la nariz es ancha y plana (Fig. 8.18)..15
Los labios son mayores, la boca usualmente se
encuentra abierta con una lengua protusiva. Los dientes están
algo separados con frecuente maloclusión. Debido a la macroglosia
presentan una distancia horizontal maxilar mayor. En la mitad de los pacientes
la erupción está retrasada y el hueso presenta áreas
de destrucción. Los pacientes pueden presentar hiperplasia gingival
debida a la pobre higiene y a la respiración bucal (Fig. 8.19).15
El retraso mental es manifiesto. El deterioro
clínico progresa rápidamente a partir del segundo o tercer
año de vida. Estos niños quedan inmovilizados, con una
creciente rigidez y contractura de las articulaciones; generalmente fallecen
antes de los 10 años de edad como consecuencia de una pneumonia
y una falla cardiaca.15